
Масово-паралельне секвенування чи масивно-паралельне секвенування (англ. Massively Parallel Sequencing, MPS), або секвенування наступного покоління (англ. Next-Generation Sequencing, NGS), або – це уніфікована назва групи високотехнологічних методів визначення первинної нуклеотидної послідовності ДНК та РНК. Головна відмінність NGS від класичних молекулярно-біологічних підходів полягає у здатності одночасно проводити мільйони паралельних реакцій зчитування в межах однієї проточної комірки. Це забезпечило експоненційне зростання пропускної здатності приладів за одночасного зниження вартості аналізу одного нуклеотиду на кілька порядків.
Джерело: ResearchGate
1. Історичний контекст та еволюція методів геномного аналізу
Розвиток геноміки прийнято ділити на кілька технологічних етапів (поколінь), кожен з яких характеризувався зміною базової фізико-хімічної парадигми детекції молекул.
Перше покоління: Метод термінації ланцюга (Sanger Sequencing)
Розроблений Фредеріком Сенгером у 1977 році метод дидезоксинуклеотидної термінації став першим золотим стандартом молекулярної біології. Реакція базується на використанні ДНК-полімерази, класичних дезоксинуклеотидтрифосфатів (dNTPs) та модифікованих дидезоксинуклеотидтрифосфатів (ddNTPs), які позбавлені 3′-ОН групи. Включення ddNTP у синтезований ланцюг унеможливлює формування наступного фосфодіефірного зв’язку, що призводить до зупинки (термінації) реплікації.
Еволюція капілярного електрофорезу з лазерною детекцією флуоресцентних міток дозволила автоматизувати процес, що зробило можливим реалізацію міжнародного проєкту «Геном людини» (Human Genome Project, HGP). Проєкт, що тривав з 1990 по 2003 рік, вимагав залучення десятків світових консорціумів, коштував близько 2.7 мільярдів доларів і дозволив прочитати лише один збірний референсний геном. Обмеженнями першого покоління є капілярний принцип (один капіляр – один фрагмент ДНК у конкретний проміжок часу), ліміт довжини читання (до 800–1000 пар основ) та висока питома вартість.
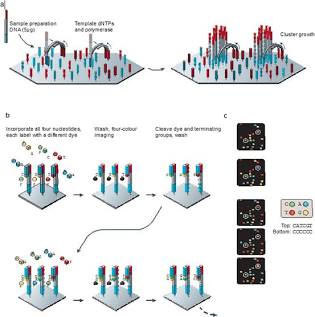
Друге покоління: Масово-паралельний синтез (MPS, NGS)
Комплаєнтний прорив відбувся у 2005 році з виходом на ринок системи GS 20 від компанії 454 Life Sciences (піросеквенування). Замість розділення фрагментів за розміром в електрофорезному полі, нові технології запропонували фіксацію молекул ДНК на твердій фазі (твердій підкладці) з їхньою одночасною ампліфікацією та детекцією сигналу безпосередньо у процесі покрокового включення нуклеотидів (Sequencing-by-Synthesis, SBS).
Це нівелювало етап індивідуального капілярного розділення та бактеріального клонування фрагментів. Вартість повногеномного дослідження знизилася за законом, що значно випереджає відомий у мікроелектроніці закон Мура.
2. Етапи технологічного процесу NGS (Workflow)
Сучасний робочий процес масово-паралельного секвенування другого покоління стандартизований і складається з трьох послідовних макроетапів: пре-аналітичного (підготовка бібліотек), аналітичного (ампліфікація та оптична/напівпровідникова детекція) та пост-аналітичного (біоінформатична обробка).
[Виділення ДНК/РНК] ──> [Фрагментація] ──> [Лігація адаптерів] ──> [Клональна ампліфікація] ──> [Циклічний синтез і детекція]
Підготовка бібліотек (Library Preparation)
Якість вихідної ДНК/РНК (цілісність, відсутність інгібіторів ПЛР) визначає успіх усього дослідження. На етапі підготовки біологічна ДНК перетворюється на стандартизовану колекцію фрагментів, придатну для взаємодії з детектуючими системами приладу.
- Фрагментація: Вихідна високомолекулярна ДНК розчіплюється на фрагменти заданої довжини (зазвичай від 150 до 500 пар основ). Використовують фізичні методи (ультразвукова кавітація на приладах Covaris), ферментативні методи (рестриктази або неспецифічні ендонуклеази) або тагментацію (використання транспозази Tn5, яка одночасно розщеплює ДНК і приєднує до її кінців олігонуклеотидні послідовності).
- Репарація кінців та А-хвостування (End Repair & A-tailing): Фрагменти після фізичного розщеплення мають нерівні кінці. За допомогою полімераз та екзонуклеаз кінці вирівнюються (створюються тупі кінці), а на 3′-кінці додається один неспарений аденін (А). Це необхідно для специфічного зв’язування з Т-основою адаптера.
- Лігація адаптерів: До модифікованих кінців фрагментів за допомогою ДНК-лігази приєднуються синтетичні дволанцюжкові олігонуклеотиди – адаптери. Вони виконують три функції:
- Праймінг: Містять послідовності, комплементарні олігонуклеотидам на проточній комірці, що дозволяє зафіксувати ДНК.
- Індексація (Баркодування): Містять унікальні послідовності нуклеотидів (6–10 баз), що виступають як цифрові паспорти зразків. Це дозволяє змішувати ДНК десятків різних пацієнтів в одній пробірці (мультиплексування) і секвенувати їх одночасно, розділяючи дані на етапі біоінформатичного аналізу.
- Унікальні молекулярні ідентифікатори (UMI): Використовуються для маркування кожної початкової молекули, що дозволяє відрізнити реальні ультрарідкісні мутації від артефактів, які виникли під час ПЛР.
Клональна ампліфікація (Template Amplification)
Одиничний флуоресцентний сигнал від одного нуклеотиду занадто слабкий для реєстрації оптичними датчиками сучасних приладів. Для подолання шумового бар’єра молекули бібліотеки клонують у просторово ізольованих точках.
- Місткова ампліфікація (Bridge Amplification – Illumina): Одноланцюжкові фрагменти ДНК з адаптерами пропускаються крізь проточну комірку, поверхня якої щільно вкрита двома типами олігонуклеотидів. Фрагмент ДНК зв’язується з першим типом олігонуклеотиду, згинається в дугу (міст) і гібридизується з другим типом олігонуклеотиду. Полімераза синтезує комплементарний ланцюг. Процес денатурації та реплікації повторюється циклічно, формуючи щільний просторовий кластер, який містить близько 1000 ідентичних копій початкової молекули (так звану полонію).
- Емульсійна ПЛР (emPCR – Ion Torrent): Реакційна суміш, що містить ДНК-бібліотеку, ПЛР-компоненти та спеціальні магнітні мікросфери, вкриті олігонуклеотидами, збовтується в мінеральній оливі. Утворюється емульсія типу «вода в оливі», де кожна мікрокрапля діє як індивідуальний мікрореактор. Усередині краплі поодинока ДНК-молекула ампліфікується безпосередньо на поверхні мікросфери, утворюючи мільйони копій. Далі мікросфери механічно розподіляються по мікролунках напівпровідникового чипа.
- ДНК-нанокулі (DNA Nanoballs – MGI / BGI): Одноланцюжкові фрагменти ДНК лігуються у кільцеві структури. За допомогою бактеріофагової полімерази Phi29 запускається процес реплікації за принципом «ротаційного кола» (Rolling Circle Amplification, RCA). Протягом кількох хвилин синтезується довгий безперервний лінійний ланцюг, що містить сотні тандемних повторів вихідного фрагмента. Завдяки фізико-хімічним властивостям цей ланцюг самоорганізується у щільну сферичну структуру – ДНК-нанокулю (DNB) діаметром близько 200 нм. Нанокулі адсорбуються на регулярній наноструктурованій матриці чипа, де кожна куля займає чітко одну позицію, що виключає проблему оптичного накладання сигналів.
3. Провідні комерційні технології другого покоління
| Платформа (Виробник) | Метод ампліфікації | Принцип детекції сигналу | Характерна довжина читання (Reads) |
| Illumina (HiSeq, NovaSeq, MiSeq) | Місткова ампліфікація на проточній комірці | Оптичний: циклічна фіксація флуоресценції чотирьох кольорів/двох каналів з оборотними термінаторами (SBS) | 2 × 75 до 2 × 300 пар основ |
| Ion Torrent (Thermo Fisher) | Емульсійна ПЛР (emPCR) на мікросферах | Напівпровідниковий: реєстрація зміни pH (іони $H^+$) при приєднанні природних нуклеотидів | 200 – 400 пар основ |
| MGI / DNBSEQ (BGI Group) | ДНК-нанокулі (DNA Nanoballs) за методом RCA | Оптичний: високошвидкісна флуоресцентна детекція за технологією cPAS (combinatorial Probe-Anchor Synthesis) | 2 × 100 до 2 × 200 пар основ |
4. Третє покоління: Секвенування поодиноких молекул у реальному часі (Long-Read Sequencing)
Головним недоліком другого покоління є необхідність збирання геному з коротких шматочків (Short Reads). Це унеможливлює точний аналіз довгих повторів, псевдогенів, мобільних генетичних елементів та великих структурних перебудов хромосом. Третє покоління вирішило цю проблему, відмовившись від ампліфікації.
Технологія SMRT (Single-Molecule Real-Time) від Pacific Biosciences (PacBio)
Метод базується на використанні субмікронних металевих структур – хвилеводів нульової моди (Zero-Mode Waveguides, ZMW), які вигравірувані на поверхні чипа. На дні кожного ZMW закріплена одна молекула ДНК-полімерази. Об’єм ZMW настільки малий, що лазерний промінь підсвічує лише нижню зону, де працює фермент.
При включенні нуклеотиду, міченого флуорофором на фосфатній групі (а не на основі, як в Illumina), прилад реєструє спалах світла. Полімераза сама відщеплює мітку в процесі формування зв’язку, що дозволяє вести безперервний синтез без зупинок на деблокування. Довжина зчитувань PacBio сягає понад 50 000 – 100 000 пар основ із високою точністю в режимі HiFi (циклічне читання замкненого кільця ДНК).
Нанопорове секвенування (Oxford Nanopore Technologies – ONT)
Принцип ONT докорінно відрізняється від оптичних систем. У синтетичну полімерну мембрану, що розділяє дві камери з електролітом, вмонтовані білкові нанопори (альфа-гемолізин або CsgG). Через мембрану пропускають постійний електричний струм, створюючи потік іонів через пору. Молекула ДНК, розплетена моторним білком-геліказою, протягується крізь нанопору.
Кожен нуклеотид або комбінація з кількох основ (к-мер), знаходячись у звуженні пори, закриває її просвіт унікальним чином, викликаючи специфічний ступінь блокування іонного струму. Амперметр високої чутливості фіксує коливання сили струму в реальному часі, а нейромережі декодують ці графіки (іонні треки) у літерну послідовність А, Т, Г, Ц. Технологія ONT дозволяє читати ультрадовгі фрагменти (рекорд перевищує 4 мільйони пар основ), аналізувати пряму РНК без переведення в кДНК та визначати епігенетичні модифікації (метилювання ДНК) безпосередньо під час секвенування.
5. Біоінформатичний аналіз: від первинних сигналів до клінічного висновку
Сирий вихід секвенатора – це файли інтенсивності флуоресценції або коливань струму. Перетворення цих даних на зрозумілий медичний звіт вимагає виконання складного біоінформатичного конвеєра (Pipeline).
[FASTQ] (Сирі дані + Якість Q) ──> [BAM/SAM] (Картування на референс) ──> [VCF] (Пошук варіантів) ──> [Медична анотація]
1) Первинний аналіз (Basecalling & Quality Control): Перетворення фізичних сигналів у текстовий формат. Стандартом є формат FASTQ, де для кожного нуклеотиду прописується бал якості Phred (Quality Score, Q). Формула розрахунку балу якості має логарифмічний вигляд:
де P – ймовірність помилкового визначення основи. Значення Q30 вказує на точність 99.9% (ймовірність помилки 1 на 1000), що є мінімальним критерієм для клінічної діагностики.
2) Вторинний аналіз (Alignment / Mapping): Отримані мільйони коротких зчитувань порівнюють із еталонним референсним геномом людини (актуальні версії GRCh38 або T2T-CHM13) за допомогою алгоритмів вирівнювання (наприклад, BWA або Bowtie2). Результат зберігається у файлах форматів SAM або його бінарній (стислій) версії BAM.
3) Пошук генетичних варіантів (Variant Calling): Спеціалізовані програмні пакети (GATK, FreeBayes) сканують BAM-файл на наявність статистично значущих відхилень від референсу. Вони фіксують:
- SNV (Single Nucleotide Variants): Однонуклеотидні заміни (точні мутації).
- Indels: Малі вставки та делеції розміром до 50 пар основ.
- CNV (Copy Number Variations): Зміни кількості копій ділянок ДНК (делеції та дуплікації великих сегментів генів).Вихідний файл цього етапу має формат VCF (Variant Call Format).
6. Клінічні стратегії застосування NGS
Залежно від діагностичного завдання та економічної доцільності, у сучасній медицині застосовують три базові рівні дослідження геному.
Таргетні клінічні панелі (Targeted Gene Panels)
Секвенуванню підлягає суворо визначений набір генів (від кількох одиниць до кількох сотень), які мають доведену клінічну асоціацію з конкретною патологією.
- Переваги: Надвисока глибина покриття ( частіше більше $500\times$ – $1000\times$), що дозволяє детектувати мозаїчні мутації; низька ціна; мінімальний ризик виявлення знахідок невідомого клінічного значення (VUS).
- Застосування: Панелі «Спадковий рак молочної залози та яєчників» (BRCA1, BRCA2, TP53 тощо), кардіоміопатії, панелі спадкових епілепсій у дітей.
Повноекзомне секвенування (Whole Exome Sequencing, WES)
Екзом – це сукупність усіх екзонів (кодуючих ділянок генів) у геномі людини. Він становить усього 1.5–2% від загального об’єму ДНК (близько 30–40 мільйонів пар основ), проте саме тут зосереджено близько 85% усіх відомих патогенних мутацій, відповідальних за спадкові хвороби.
- Переваги: Універсальний інструмент для пошуку причин рідкісних, атипових або орфанних захворювань, коли клінічна картина розмита.
- Застосування: Подолання «діагностичної одіссеї» у дітей із затримками психомоторного розвитку, вродженими вадами чи імунодефіцитними станами нез’ясованого генезу.
Повногеномне секвенування (Whole Genome Sequencing, WGS)
Аналіз абсолютно всієї послідовності ДНК, включаючи екзони, інтрони, міжгенні проміжки, промоторні ділянки та мітохондріальний геном.
- Переваги: Максимально повна інформація. WGS дозволяє виявляти структурні варіанти хромосом, глибокі інтронна мутації, що впливають на сплайсинг, та перебудови, які пропускає екзомне секвенування. Забезпечує фундаментальний профіль здоров’я на все життя.
7. Критерії інтерпретації генетичних варіантів (Стандарти ACMG)
Виявлення мутації у файлі VCF не означає автоматичного встановлення діагнозу. Відповідно до міжнародних консенсусних рекомендацій Американської колегії медичної генетики та геноміки (ACMG) та Асоціації молекулярної патології (AMP), кожен знайдений генетичний варіант класифікується за п’ятирівневою шкалою шкідливості:
- Class 1: Pathogenic (Патогенний) – Зв’язок мутації із хворобою доведений, функція білка повністю порушена.
- Class 2: Likely Pathogenic (Ймовірно патогенний) – З високою ймовірністю (більше 90%) варіант є причиною патології.
- Class 3: Variant of Uncertain Significance (VUS – Варіант із невизначеним клінічним значенням) – Недостатньо наукових даних, біоінформатичні предиктори дають суперечливі прогнози. Вимагає спостереження та тестування родичів (сегрегаційний аналіз).
- Class 4: Likely Benign (Ймовірно доброякісний) – Найімовірніше є безпечним поліморфізмом.
- Class 5: Benign (Доброякісний) – Варіант є нормою для популяції (природна мінливість).
8. Забезпечення світових стандартів тестування
Організація молекулярно-генетичного аналізу класу NGS для пацієнтів в Україні через сервіс ГенТест базується на принципах суворої доказовості та оптимізації кожного етапу логістики біоматеріалу. Сервіс виступає інформаційно-логістичним партнером лідерів світового ринку – лабораторій Invitae (США) та Blueprint Genetics (Фінляндія).
Неінвазивна стабілізація біоматеріалу
Замість класичного забору венозної крові використовуються спеціалізовані сертифіковані набори з консервуючим буфером. Пацієнт здає зразок слини або букального епітелію (мазок з унутрішньої сторони щоки).
Хімічний склад буфера миттєво лізує клітини епітелію та інактивує нуклеази (ферменти, що розщеплюють ДНК), фіксуючи та стабілізуючи молекули ДНК. У такому стані ДНК залишається стабільною при температурах від мінус 20 до плюс 50 градусів Цельсія протягом декількох тижнів, що виключає ризик деградації матеріалу в дорозі.
Транспортна та митна логістика (Cold/Ambient Chain)
Транспортування зразків з України до лабораторій у США або ЄС здійснюється експрес-службами міжнародної доставки (FedEx, DHL, UPS) за регламентом перевезення біологічних матеріалів категорії B (UN 3373). Наш сервіс бере на себе повний цикл митного оформлення, що гарантує проходження кордонів без затримок на митних складах, забезпечуючи дотримання часових лімітів лабораторій.
Верифікація у лабораторіях за стандартами CLIA та CAP
Всі аналізи виконуються на базі лабораторій, що мають акредитацію САР (College of American Pathologists) та сертифікацію CLIA (Clinical Laboratory Improvement Amendments). Це гарантує, що:
- Точність детекції варіантів верифікована незалежними панелями якості.
- Біоінформатична обробка використовує регулярно оновлювані бази даних клінічних варіантів (ClinVar, gnomAD).
- Фінальний звіт підписується сертифікованим медичним генетиком США або ЄС.
Автор матеріалу:
Степан Бегларян, MD, PhD,
дитячий імунолог, педіатр
Список джерел
- Heather, J. M., & Chain, B. (2016). The sequence of sequencers: The history of sequencing DNA. Genomics, 107(1), 1–8
- Goodwin, S., McPherson, J. D., & McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics, 17(6), 333–351
- Kchouk, M., Gibrat, J. F., & Elloumi, M. (2017). Generations of Sequencing Technologies: From First to Next Generation. Biology and Medicine (Aligarh), 9(3), 395
- Metzker, M. L. (2010). Sequencing technologies — the next generation. Nature Reviews Genetics, 11(1), 31–46
- Ansorge, W. J. (2009). Next-generation DNA sequencing techniques. New Biotechnology, 25(4), 195–203
- Mardis, E. R. (2011). A decade’s perspective on DNA sequencing technology. Nature, 470(7333), 198–203
- Shendure, J., & Ji, H. (2008). Next-generation DNA sequencing. Nature Biotechnology, 26(10), 1135–1145
- Voelkerding, K. V., Dames, S. A., & Durtschi, J. D. (2009). Next-Generation Sequencing: From Basic Research to Diagnostics. Clinical Chemistry, 55(4), 641–658
- Sanger, F., Nicklen, S., & Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences, 74(12), 5463–5467
- Margulies, M., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature, 437(7057), 376–380
- Bentley, D. R., et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature, 456(7218), 53–59
- Rothberg, J. M., et al. (2011). An integrated semiconductor device enabling non-optical genome sequencing. Nature, 475(7356), 348–352
- Eid, J., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science, 323(5910), 133–138